
Cytochrome P450 (CYP)
 Pharmakokinetik MetabolismusDie Cytochrome P450 (CYP) sind eine Familie von Enzymen, die für den Metabolismus der Arzneimittel von zentraler Bedeutung sind. Wichtige Mitglieder sind beispielsweise CYP2B6, CYP2C9, CYP2C19, CYP2D6 und CYP3A. Pharmazeutische Wirkstoffe, die von CYP-Isoenzymen verstoffwechselt werden, sind anfällig für Arzneimittel-Wechselwirkungen. Denn andere Medikamente können die Enzyme hemmen oder ihre Expression erhöhen. Dadurch steigt das Risiko für unerwünschte Wirkungen oder für einen Wirkungsverlust. CYP450
Pharmakokinetik MetabolismusDie Cytochrome P450 (CYP) sind eine Familie von Enzymen, die für den Metabolismus der Arzneimittel von zentraler Bedeutung sind. Wichtige Mitglieder sind beispielsweise CYP2B6, CYP2C9, CYP2C19, CYP2D6 und CYP3A. Pharmazeutische Wirkstoffe, die von CYP-Isoenzymen verstoffwechselt werden, sind anfällig für Arzneimittel-Wechselwirkungen. Denn andere Medikamente können die Enzyme hemmen oder ihre Expression erhöhen. Dadurch steigt das Risiko für unerwünschte Wirkungen oder für einen Wirkungsverlust. CYP450Die Cytochrome P450 sind eine Familie von Enzymen, die für die Biotransformation von Medikamenten von herausragender Bedeutung sind.
Die wichtigsten Isoenzyme für den Arzneistoffmetabolismus sind:
- CYP1A1, CYP1A2
- CYP2B6
- CYP2C9, CYP2C19
- CYP2D6
- CYP2E1
- CYP3A4, CYP3A5 und CYP3A7
Die Zahl nach der Abkürzung CYP steht für die Familie, der darauffolgende Buchstabe für die Unterfamilie und die letzte Nummer für das einzelne Enzym.
Die Cytochrome sind vorwiegend in der Leber lokalisiert, sie kommen aber auch in anderen Organen vor, insbesondere im Darm. Sie spielen eine wichtige Rolle für den First-Pass-Metabolismus.
Einige Familienmitglieder sind auch am Stoffwechsel von körpereigenen Molekülen wie Steroiden, Gallensäuren, Fettsäuren, Eicosaniden und fettlöslichen Vitaminen beteiligt.
Chemische ReaktionenCYPs enthalten als Cofaktor ein Molekül Häm im Protein. Mit dem zentralen Eisenatom binden und aktivieren sie Sauerstoff und übertragen je ein Atom Sauerstoff auf das Substrat und ein neu gebildetes Wassermolekül. Die Enzyme werden deshalb als Monooxygenasen bezeichnet. Sie katalysieren die folgende allgemeine Reaktion, wobei R-H für das Substrat steht:
R–H + O2 + NADPH + H+ → R–OH + H2O + NADP+
Diese Reaktion entspricht einer Hydroxylierung. Es entsteht ein Alkohol oder bei einer aromatischen Hydroxylierung ein Phenol. So wird beispielsweise das Schmerzmittel Ibuprofen von CYP2C9 hydroxyliert:
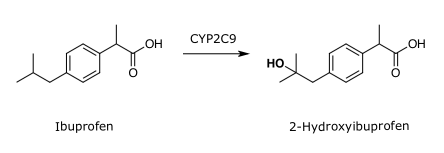
Weitere Reaktionen sind eine Epoxidierung, eine Dealkylierung, eine Desaminierung und eine Oxidation.
Die Veränderung der chemischen Struktur eines Moleküls kann zu einer Aktivierung oder einer Inaktivierung führen. Einige Gifte - wie beispielsweise Aflatoxin B1 - sind Prodrugs und werden erst von den Cytochromen toxifiziert.
Arzneimittel-WechselwirkungenCYP-Substrate sind anfällig für Arzneimittel-Wechselwirkungen mit Inhibitoren oder Induktoren der metabolischen Enzyme. Besonders bei Substanzen mit einer engen therapeutischen Breite, mit einer hohen Toxizität und Arzneimitteln, welche das QT-Intervall verlängern, ist Vorsicht geboten.
CYP-InhibitorenCYP-Inhibitoren sind Wirkstoffe oder andere Substanzen, welche die Aktivität von CYP-Isoenzymen reduzieren. Dies führt beispielsweise dazu, dass die Inaktivierung eines Wirkstoffs vermindert wird. Dadurch steigt das Risiko für unerwünschte Wirkungen.
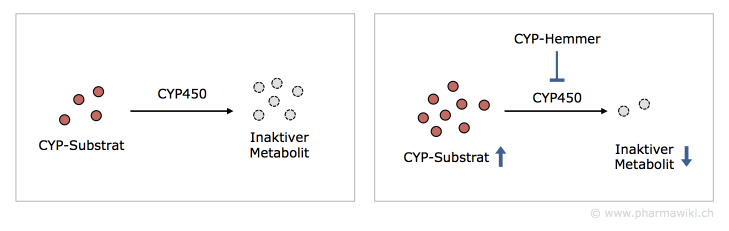
Sind CYP450-Isoenzyme an der Aktivierung eines Prodrugs beteiligt, hat eine Hemmung zur Folge, dass weniger aktiver Wirkstoff entsteht. Es kann zu einem Wirkungsverlust kommen.
Bekannte CYP-Inhibitoren sind die Azol-Antimykotika, Makrolide (häufig: Clarithromycin) und HIV-Proteasehemmer.
CYP-InduktorenCYP-Induktoren erhöhen die Enzymaktivität, indem sie die Proteinsynthese stimulieren. Der Effekt tritt mit einer zeitlichen Verzögerung ein. Dadurch werden die CYP-Substrate verstärkt metabolisiert. Dies hat beispielsweise zur Folge, dass Ethinylestradiol, das Östrogen in vielen hormonalen Verhütungsmitteln, verstärkt abgebaut wird und es zu einer Schwangerschaft kommt.
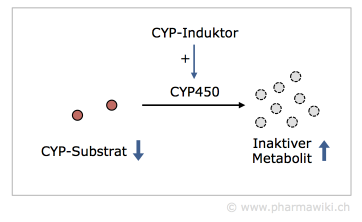
Typische Beispiele sind die Rifamycine wie Rifampicin, die Barbiturate, Johanniskraut und Antiepileptika wie Carbamazepin.
Pharmakokinetische BoosterDie oben beschriebenen Arzneimittel-Wechselwirkungen sind nicht zwangsläufig unerwünscht. Pharmakokinetische Booster wie Ritonavir oder Cobicistat werden mit CYP-Substraten kombiniert, um ihren Abbau zu bremsen und die Bioverfügbarkeit zu erhöhen. Bei den Boostern handelt es sich in der Regel um CYP-Hemmer.
PharmakogenetikDie Enzymaktivität der Cytochrome ist aufgrund der Genetik interindividuell verschieden. Dies trifft insbesondere auf CYP2D6 und CYP2C19 zu. In der Bevölkerung existieren schnelle, langsame und normale Metabolisierer.
Bei CYP2D6 sind bis zu 15% der Bevölkerung langsame Metabolisierer (!) Ein typisches CYP2D6-Substrat ist das Antitussivum Dextromethorphan. Ein bekanntes Substrat von CYP2C19 ist Clopidogrel, das von diesem Enzym aktiviert wird.
Der Effekt auf die Pharmakokinetik ist mit einer CYP-Inhibition oder CYP-Induktion vergleichbar. Langsame Metabolisierer haben ein erhöhtes Risiko für unerwünschte Wirkungen, bei schnellen Metabolisieren kann der Effekt eines Medikaments ausbleiben.
Heute ist es möglich, das genetische Profil mit einer Laboranalyse einfach zu bestimmen und Therapien zu individualisieren. Dadurch lassen sich Nebenwirkungen vermeiden.
Reaktion auf Wechselwirkungen- Beurteilung der klinischen Relevanz, z.B. mithilfe der Fachinformation oder mit Applikationen
- Vorübergehendes oder dauerhaftes Absetzen eines Medikaments
- Wechsel eines Medikaments
- Dosisanpassungen
Interaktionen, Metabolismus, Glucuronidierung
Literatur- Arzneimittel-Fachinformation (CH)
- Backman J.T., Filppula A.M., Niemi M., Neuvonen P.J. Role of Cytochrome P450 2C8 in Drug Metabolism and Interactions. Pharmacol Rev, 2016, 68(1), 168-241 Pubmed

- Conner K.P., Woods C.M., Atkins W.M. Interactions of cytochrome P450s with their ligands. Arch Biochem Biophys, 2011, 507(1), 56-65 Pubmed
- Evers R. et al. Critical review of preclinical approaches to investigate cytochrome p450-mediated therapeutic protein drug-drug interactions and recommendations for best practices: a white paper. Drug Metab Dispos, 2013, 41(9), 1598-609 Pubmed
- Galetin A., Gertz M., Houston J.B. Contribution of intestinal cytochrome p450-mediated metabolism to drug-drug inhibition and induction interactions. Drug Metab Pharmacokinet, 2010, 25(1), 28-47 Pubmed
- Guengerich F.P. Cytochromes P450, drugs, and diseases. Mol Interv, 2003, 3(4), 194-204 Pubmed
- Guengerich F.P. Cytochrome P450s and other enzymes in drug metabolism and toxicity. AAPS J, 2006, 8(1), E101-11 Pubmed
- Lynch T., Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician, 2007, 76(3), 391-6 Pubmed
- Mazaleuskaya L.L. et al. PharmGKB summary: ibuprofen pathways. Pharmacogenet Genomics, 2015, 25(2), 96-106 Pubmed
- Vögtli A., Ernst B. Moderne Pharmakokinetik. Transport durch Membranen. Weinheim: Wiley-VCH, 2010
- Wroblewski B., Glenn M.B. The cytochrome p-450 drug metabolizing enzyme system: an overview of potential clinically important drug interactions. J Head Trauma Rehabil, 2002, 17(6), 571-4 Pubmed
- Zanger U.M., Raimundo S., Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Arch Pharmacol. 2004, 369(1), 23-37 Pubmed
Interessenkonflikte: Keine / unabhängig. Der Autor hat keine Beziehungen zu den Herstellern und ist nicht am Verkauf der erwähnten Produkte beteiligt.